
科学网
据英国每日邮报报道,科学家和心理学家长期以来一直未解开一个谜团——为什么我们会忘记童年发生的事情?这是人生之中最无忧无虑,最快乐的时光。目前,两位科学家认为他们找到了揭开谜底的答案,指出生命初期大脑记忆中枢的细胞快速生长,意味着早期存在的大脑记忆细胞之间的关键性连接将被更新替代,因此,人们童年时期的记忆就不太可能恢复。
 在加拿大神经系统科学会议上,专家们听取了关于为什么婴儿仅能记忆生日宴会事件几天时间的理论解释。相比之下,年龄稍大的儿童和成年人大脑细胞之间的连接就变得更加稳定,使记忆能够很好地存储下来。
在加拿大神经系统科学会议上,专家们听取了关于为什么婴儿仅能记忆生日宴会事件几天时间的理论解释。相比之下,年龄稍大的儿童和成年人大脑细胞之间的连接就变得更加稳定,使记忆能够很好地存储下来。
这一理论是由一对夫妻科学家提出的,他们是来自加拿大多伦多大学的保罗-弗兰克德(Paul Frankland)和希娜-乔塞尔(Sheena Josselyn),他们指出幼年老鼠记忆中枢形成的细胞更容易使它们忘记一些记忆。同时,还发现幼年老鼠无法生成具有较好记忆的正常记忆细胞。
之前研究表明,我们无法回忆起2-3岁之前的事情,只能在3-7岁之间产生一些零碎的回忆。该现象的解释有诸多因素,其中包括:记忆随时间而模糊消退,记忆形式与语言能力的形式紧密相联。多年以来,婴儿健忘症一直是一个难解的科学谜团,目前弗兰克德夫妇认为他们的解释理论具有一定的说服力。
弗兰克德称,我们的最新研究解释了为什么人类幼年时期很少有回忆,同时,无法回忆起童年生活的细节也具有一定的好处。例如:他们两岁大的女儿在生日蛋糕蜡烛燃灭时突然啼哭。他们指出,这个事件会在我们的记忆中永久地留下印记,我们能够生动地回忆着生日庆祝中不愉快的事情,我们的女儿也会记忆这件事,但只有一两天的记忆,第二天她会大叫“没有蜡烛”,用力地摆动自己的头。我们何必让女儿的回忆伤痕累累呢?婴儿健忘症会让他们忘记童年的一些事情,当女儿成年时,不会清晰地回忆两岁生日时的“蜡烛事件”。
关键词:
雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)是一种丝氨酸/苏氨酸蛋白激酶,在细胞生长、分化、增殖、迁移和存活上扮演重要角色。雷帕霉素常用于组织移植排斥,先前有研究证实TOR是雷帕霉素的靶标,后来又有发现TOR有抗癌效果。2002年在酵母中发现的雷帕霉素不敏感的TOR信号途径(rapamycin-insensitive TOR pathway),并没有将此蛋白革名,反而使人们更加确信mTOR存在于两种完全不同的多蛋白复合体—— mTORC1和mTORC2中。 最近两篇热点封面介绍说两个实验室发现一种哺乳动物mTORC2,即雷帕霉素不敏感复合体。
参考链接:
http://www.nature.com/neuro/journal/v16/n4/full/nn.3351.html
-----------------
mtorc2: actin on your memory
Sheena A Josselyn & Paul W Frankland
To be become long-lasting, short-term memories must be transformed into more permanent forms. mTORC2 has now been found to be crucial for the molecular reorganization of the cytoskeleton needed for memory consolidation.
Sheena A. Josselyn and Paul W. Frankland are in the Program in Neurosciences & Mental Health at The Hospital for Sick Children, and in the Departments of Psychology and Physiology and the Institute of Medical Science, University of Toronto, Toronto, Ontario, Canada.
e-mail: sheena.josselyn@sickkids.ca
How do you make a memory? The formation of a long-term memory is thought to involve at least two ingredients. First, it requires the strengthening of synaptic connections between neurons, a process that depends on protein synthesis. Second, it also requires changes in the physical structure of the synaptic connections, a process that depends on actin cytoskeleton rearrangement. Most studies have focused on the role of protein synthesis in memory formation; much less is known about actin polymerization and memory. Although protein synthesis and actin cytoskeleton rearrangement must be intimately intertwined, even less is known about how these necessary memory processes interact at the molecular level. A study by Huang et al.1 in this issue of Nature Neuroscience reveals that mTORC2 (mammalian target of rapamycin complex 2) positively regulates the actin dynamics required for the formation of long-term memories (Fig. 1) and also suggests that actin polymerization is upstream of protein synthesis.
mTOR (mammalian target of rapamycin) is a serine-threonine protein kinase integral to signal transduction pathways that control cell growth and survival, translation, autophagy and cytoskeleton organization2. It serves as an intracellular sensor for energy metabolism, nutrient availability and stresses, regulating cellular and organism growth and metabolism to adapt to environmental changes. The best studied and understood function of mTOR is the regulation of protein translation. mTOR forms two distinct multiprotein complexes, which are distinguished by their accessory proteins. The mTOR complex 1 (mTORC1) contains the accessory protein raptor (regulatory-associated protein of mTOR) and is sensitive to the drug rapamycin. By contrast, mTORC2 contains rictor (rapamycininsensitive companion of mTOR) and is resistant to acute rapamycin treatment3–5. Not only do these mTOR complexes differ in their sensitivity to rapamycin, but they also show functional differences. mTORC1 is well characterized and is known to positively regulate protein translation necessary for long-term synaptic plasticity and memory formation6,7. In contrast, relatively little is known about the overall function of mTORC2, let alone its role in synaptic plasticity and memory. Hints from other species and tissues suggest that mTORC2, through its defining component rictor, controls the actin cytoskeleton5,8 and therefore might regulate some aspects of the structural plasticity required for memory formation. By targeting rictor, Huang et al.1 identified an important function of this complex in regulating both long-term synaptic plasticity and long-term memory formation.
Because rictor is important during brain development, Huang et al.1 developed a conditional knockout mouse in which rictor was deleted only from postnatal forebrain excitatory neurons. These rictor-deficient mice showed disrupted mTORC2, but intact mTORC1, activity. The authors first used these mice to examine the effects on synaptic plasticity of deleting mTORC2. Long-term potentiation (LTP) of excitatory synaptic responses is a commonly used electrophysiological correlate of memory formation. In the CA1 region of the hippocampus, weak tetanization (for example, one train at 100 Hz) induces early phase-LTP (E-LTP), which, as its name implies, decays over time. In contrast, stronger tetanization (for example, four trains at 100 Hz) induces late phase-LTP (L-LTP), a form of LTP that lasts for several hours. The conversion of E-LTP into L-LTP requires both protein synthesis and actin cytoskeletal rearrangement9,10. Actin is the main structural component of dendritic spines, and its rearrangement via polymerization is critical for L-LTP (but not E-LTP). Mice with a forebrain deletion of rictor showed normal basal synaptic transmission and E-LTP. However, this E-LTP could not be converted to L-LTP.
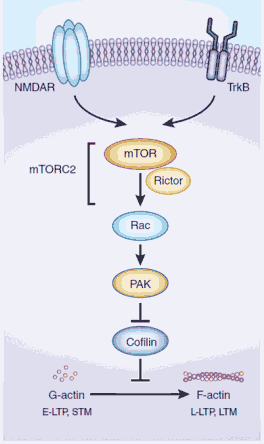
Figure 1
mTORC2 regulates actin dynamics to form long-term memories. The mTOR complex 2 (mTORC2) is composed of mTOR and rictor (rapamycin-insensitive companion of mTOR), among other accessory proteins. Several signaling pathways (for example, Ca2+ acting through NMDA-type glutamate receptors (NMDAR), or brain-derived neurotrophic factor (BDNF) acting through TrkB receptors) activate mTORC2, which in turn acts through Rac, PAK and cofilin to promote the actin polymerization required for the conversion of early LTP (E-LTP) into late LTP (L-LTP) and short-term memory (STM) into longterm memory (LTM).
Of course, studies of LTP and synaptic physiology greatly inform our understanding of the mechanistic recipe for memory formation, but the real proof is in the pudding. In short, do these mTORC2-deficient mice show impaired long-term memory? Previous studies have shown that actin polymerization is required for the conversion of a weak shortterm memory into long-term memory10. To examine mTORC2 in memory, the authors tested rictor-deficient mice in several different tasks that differ in their training schedule and performance demands. Classical fear conditioning is a memory model in which a tone is paired with a shock in a specific context. Rictor-deficient mice showed normal memory for both the context and tone when tested 2 h after training, indicating that their short-term memory was intact. However, when tested 24 h after training, they showed impaired memory for both the context and tone. These behavioral results parallel the synaptic physiology data: similarly to the failure to convert E-LTP to L-LTP, these mice seemed unable to convert a short-term memory into a long-term memory. Consistent with this conclusion, Huang et al.1 went on to examine spatial memory formation in rictor-deficient mice using the Morris water maze. In this model, mice learn to find a hidden platform submerged under the surface of an opaque liquid by using spatial cues around the room. Training takes place over several days, and therefore mice need to remember from one day to the next. Again, rictor-deficient mice were unable to form a stable spatial memory.
Many foundational studies of the molecular basis of long-term memory were conducted in invertebrates, including Drosophila melanogaster11,12. Although the results of fly studies have certainly informed rodent studies (and vice versa), these research streams have often existed in parallel universes. Notably, Huang et al.1 unite these research traditions by comparing memory in both rodents and flies with genetically disrupted mTORC2 function. Seymour Benzer and his students pioneered the use of classical fear conditioning, in which an odor is paired with shock, to examine memory in flies10. In this model, massed training (training trials with no inter-trial rest periods) produces a short-term memory that is independent of protein synthesis, whereas spaced training (training trials with intervening rest periods) produces protein synthesis–dependent long-term memory10. In a striking parallel to the findings with mice, rictor-deficient flies showed intact short-term but impaired longterm memory, indicating that the function of rictor in memory is conserved.
To better understand the mechanism behind mTORC2’s effect on memory, the authors turned to its downstream targets. Consistent with mTORC2 and rictor’s role in actin polymerization, the authors found that rictor-deficient mice showed disrupted actin dynamics. The polymerization of free globular actin (G-actin) to form filamentous actin (F-actin) is thought to underlie the growth of dendritic spines necessary for memory formation10. Rictor-deficient mice showed a reduction in the ratio of F-actin to G-actin, as well as a reduction in expression of a number of upstream positive regulators of actin polymerization. These correlational data suggested that rictor is required for long-term memory formation by increasing the F-actin important for dendritic spine growth and remodeling.To directly test this hypothesis, the authors manipulated actin itself. First they mimicked the effect of disrupting actin polymerization in otherwise normal wild-type mice using cytochalasin D, a drug that specifically disrupts actin polymerization. Like rictor-deficient slices, wild-type slices treated with cytochalasin D showed intact E-LTP but disrupted L-LTP. Is normalizing actin dynamics sufficient to rescue the L-LTP and long-term memory deficits in rictor-deficient mice? Jasplakinolide (JPK), a compound that promotes actin polymerization, restored both the F-actin to G-actin ratio and the ability to produce L-LTP in slices in rictor-deficient mice, without affecting these processes in slices from wild-type mice. Remarkably, infusion of JPK directly into the hippocampus similarly reversed the long-term memory deficits in rictor-deficient mice. Might increasing actin polymerization enhance synaptic plasticity and memory formation in wild-type mice? To examine this, the authors used weak training conditions that are normally sufficient to induce only E-LTP and short-lasting memory in wild-type mice and found that this drug enabled both L-LTP and long-term memory formation. Intriguingly, the L-LTP induced by weak training in the presence of JPK was blocked by application of anisomycin, a protein synthesis inhibitor, suggesting that actin polymerization is upstream of protein synthesis. Finally, systemic injection of a small molecule that increases mTORC2 activity (A-443654) not only facilitated L-LTP in the slice but also enhanced long-term memory formation in wild-type mice.
By both increasing and decreasing mTORC2 function, Huang et al.1 provide compelling evidence that mTORC2, acting through actin polymerization, is important in formation of stable memories. They showed that this mechanism was conserved in both flies and mice. Taken together, these findings raise the question of whether targeting mTORC2 might similarly enhance memory and other cognitive functions in humans. Given that mTORC2 disruption is implicated in many human diseases associated with cognitive dysfunction13–15, the present study offers a tantalizing prospect for future therapeutic avenues.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
1. Huang, W. et al. Nat. Neurosci. 16, 441–448 (2013).
2. Hay, N. & Sonenberg, N. Genes Dev. 18, 1926–1945 (2004).
3. Kim, D.H. et al. Cell 110, 163–175 (2002).
4. Sarbassov, D.D. et al. Curr. Biol. 14, 1296–1302 (2004).
5. Wullschleger, S., Loewith, R. & Hall, M.N. Cell 124, 471–484 (2006).
6. Tang, S.J. et al. Proc. Natl. Acad. Sci. USA 99, 467–472 (2002).
7. Stoica, L. et al. Proc. Natl. Acad. Sci. USA 108, 3791–3796 (2011).
8. Guertin, D.A. & Sabatini, D.M. Cancer Cell 12, 9–22 (2007).
9. Cingolani, L.A. & Goda, Y. Nat. Rev. Neurosci. 9, 344–356 (2008).
10. Lamprecht, R. & LeDoux, J. Nat. Rev. Neurosci. 5, 45–54 (2004).
11. Weiner, J. Time, Love, Memory: a Great Biologist and His Quest for the Origins of Behavior (Knopf, New York, 1999).
12.Dubnau, J. & Tully, T. Annu. Rev. Neurosci. 21, 407–444 (1998).
13. Murata, H. et al. J. Biol. Chem. 286, 7182–7189 (2011).
14. Ljungberg, M.C. et al. Ann. Neurol. 60, 420–429 (2006).
15.Knobbe, C.B., Trampe-Kieslich, A. & Reifenberger, G. Neuropathol. Appl. Neurobiol. 31, 486–490 (2005)
www.psychspace.com心理学空间网
